Product registration is not a checkbox you complete once before a product enters the market. For healthcare professionals and regulatory specialists managing pharmaceutical and medical device portfolios, it is an ongoing operational discipline with serious legal, commercial, and patient safety implications. Regulatory frameworks in the United States, European Union, and Singapore are all undergoing significant updates in 2026, changing timelines, system requirements, and role assignments for regulated manufacturers and distributors. This guide breaks down what those changes mean for your compliance workflows and how to stay ahead of them.
Table of Contents
- Key takeaways
- Understanding product registration under US FDA requirements
- Navigating EU requirements with EUDAMED in 2026
- Comparing registration across US, EU, and Singapore
- Best practices for managing registration and compliance data
- My perspective on registration as an operational discipline
- How Labgistics supports registration and logistics compliance
- FAQ
Key takeaways
| Point | Details |
|---|---|
| Registration is continuous | FDA and EU regulations require ongoing updates to product listings throughout the full product lifecycle. |
| EUDAMED goes mandatory in 2026 | The first four EUDAMED modules become compulsory from 28 May 2026 for all relevant medical device market participants in Europe. |
| FDA timing is strict | US device establishment registration must be renewed annually between October 1 and December 31 with no grace period. |
| Singapore updated its pathways | HSA introduced new eCTD submission requirements and GMP conformity changes effective January 2026 for therapeutic products. |
| Cross-functional alignment matters | Regulatory, quality, and supply chain teams must coordinate to prevent data inconsistencies that cause registration delays. |
Understanding product registration under US FDA requirements
The FDA frames drug and device registration as maintaining a current catalog of all products in commercial distribution. That framing is revealing. A catalog requires constant maintenance. It is not filed once and forgotten.
Who must register? The obligation applies broadly:
- Domestic drug and biologic manufacturers and processors
- Foreign establishments whose products are exported to the United States
- Repackers and relabelers of finished drug products
- Medical device manufacturers and initial importers
- Contract sterilizers and specification developers
The regulatory distinction between establishment registration and device listing is frequently misunderstood. Establishment registration identifies the physical facility and its owners. Device listing identifies the specific products manufactured at that facility that are in commercial distribution. Both must be completed within 30 days of beginning operations or commencing commercial distribution. Missing either one puts your entire US market access at risk.
FDA uses two electronic systems to manage this: FURLS (FDA Unified Registration and Listing System) for devices and DRLM (Drug Registration and Listing Module) for pharmaceutical products. Familiarity with both is non-negotiable for any regulatory specialist managing a mixed portfolio.
Annual renewal for device registrations runs from October 1 through December 31. There is no grace period after December 31. A lapsed registration can trigger import detentions and misbranding enforcement letters, both of which create immediate supply chain disruptions. For foreign establishments, an additional layer applies: you must designate a US Agent physically located in the United States. That agent serves as FDA’s primary contact for communications and inspections, but they are not authorized to register on your behalf. The regulatory accountability stays with the establishment.

Pro Tip: Set calendar alerts for October 1 every year across your regulatory and supply chain teams. By the time December approaches, renewal should already be processed. Late renewals are entirely preventable with basic scheduling discipline.
The most common pitfalls in FDA registration? Incomplete device listings that omit products added mid-year, failure to update listings after product discontinuation, and missing the renewal window due to internal communication gaps between regulatory affairs and finance teams responsible for fee payment.
Navigating EU requirements with EUDAMED in 2026
The shift happening in Europe this year is more significant than most compliance teams are treating it. The first four EUDAMED modules become mandatory starting 28 May 2026. These four modules cover:
- Actor registration: All economic operators, authorized representatives, and importers must be registered and hold a Single Registration Number (SRN).
- UDI and device registration: New devices placed on the EU market after May 2026 must have their UDI data registered before market placement.
- Notified body certificates: Certificate data must be uploaded by Notified Bodies, increasing transparency across the sector.
- Market surveillance: Vigilance data and post-market surveillance connections feed into regulatory oversight.
The SRN is not optional. Without it, device market placement cannot proceed after May 2026. If your team has not yet secured an SRN through actor registration in EUDAMED, that process needs to begin now. Registration alone can take weeks depending on the competent authority in your member state.
EUDAMED also introduces a two-actor requirement for device registration. The proposer submits device registration data and the confirmer validates it. One person cannot hold both roles. This role separation has real operational consequences for smaller regulatory teams that may need to involve a local actor administrator or authorized representative to fulfill the confirmer function.
| Requirement | Who is responsible | Deadline |
|---|---|---|
| SRN via actor registration | Economic operator, authorized representative | Before 28 May 2026 |
| UDI/device registration for new devices | Manufacturer or authorized representative | Before market placement |
| Certificate uploads | Notified Body | Upon certification |
| Vigilance data linking | Manufacturer | Post-market phase |
Legacy devices already on the market face a transition timeline tied to their MDR certification dates. But new devices have no flexibility. The EUDAMED mandatory modules apply without exception from May 28.
A frequently overlooked implication: EUDAMED’s notified body certificate module makes competitor certification data publicly accessible. Your regulatory status and certificate timelines are now visible to competitors, regulators, and procurement teams in a way they were not before. That transparency cuts both ways.
Pro Tip: Before 28 May 2026, run an internal QA gate that cross-checks your labeling, IFU content, and UDI data against your EUDAMED submissions. Labeling and UDI consistency is one of the highest-frequency causes of registration delays, and fixing it before submission is far less costly than correcting it post-submission.
Comparing registration across US, EU, and Singapore
Understanding how registration workflows differ by region lets your team allocate resources and set realistic timelines. Here is a structured comparison of the three major regulatory environments relevant to most healthcare companies operating in Asia-Pacific.
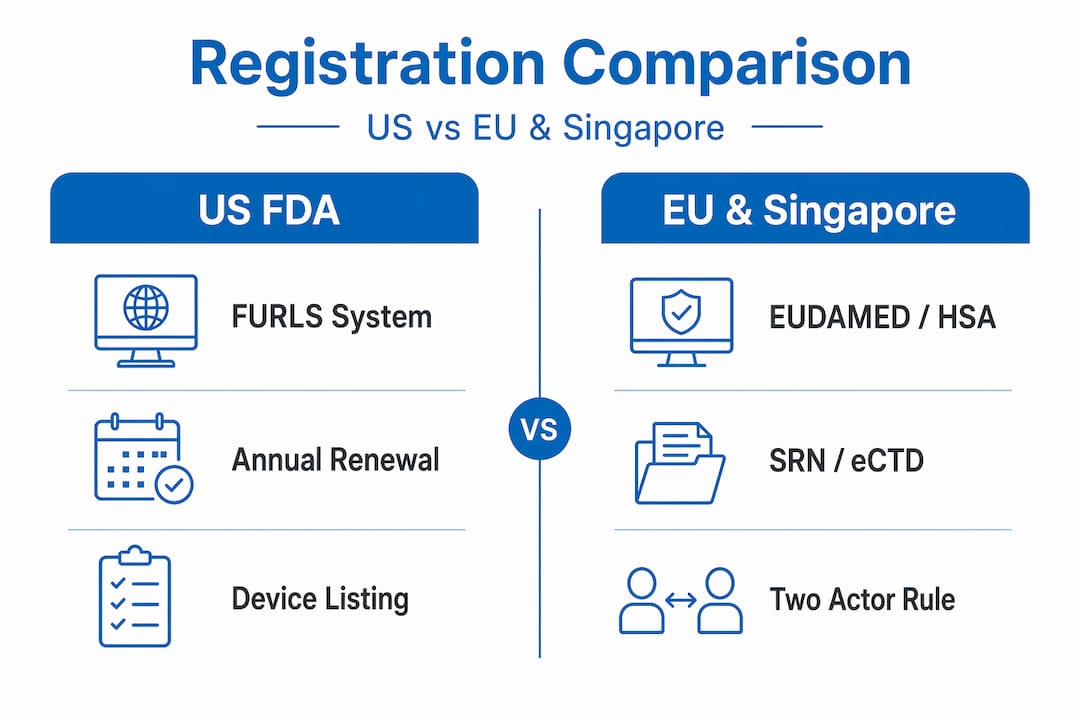
| Regulatory dimension | US FDA | EU EUDAMED | Singapore HSA |
|---|---|---|---|
| Primary system | FURLS / DRLM | EUDAMED | PRISM / eCTD |
| Registration type | Establishment + product listing | Actor + UDI/device registration | Product license application |
| Mandatory renewal | Annual (Oct 1 to Dec 31) | Certificate-linked | License term-based renewal |
| Electronic submission | Required | Required from May 2026 | eCTD from Jan 2026 |
| Foreign entity requirement | US Agent required | Authorized representative required | Local agent or liaison required |
Singapore’s 2026 regulatory updates) introduced a test phase for eCTD submissions for therapeutic products and new GMP conformity assessment requirements for overseas manufacturing sites. For companies registering pharmaceutical products in Singapore, these changes affect both the submission format and the evidence package required for overseas facility oversight.
For teams looking at product registration in Singapore, the HSA pathway for therapeutic products operates under a risk-based classification framework, with full registration, abridged, or verification pathways depending on the product’s regulatory history in reference markets. Aligning your Singapore submission with existing FDA or EMA approval data can significantly reduce review timelines.
Across all three markets, certain principles hold constant:
- UDI compliance must be reflected in both regulatory submissions and physical labeling
- Regulatory submissions must align precisely with the technical file or dossier content
- Product discontinuation or reformulation triggers mandatory updates in all systems
- Supply chain partners, including 3PL providers, need current registration information to manage import and distribution documentation correctly
Best practices for managing registration and compliance data
The most expensive compliance failures in product registration share a common root cause: data that is accurate in one system but outdated or inconsistent in another. A device listing filed with FDA that reflects an old labeling version while the physical product carries updated labeling is a real enforcement vulnerability, regardless of intent.
Managing registration data well requires:
- A single source of truth. Whether you use a regulatory information management system (RIMS) or structured document control, every registration entry must trace back to the same approved technical documentation.
- Scheduled review cycles. Do not wait for a renewal deadline to discover that a product has been reformulated or relabeled. Quarterly reviews of active registrations against current product status catch drift early.
- Cross-functional handoffs. When the quality team approves a labeling change, that approval must automatically trigger a regulatory review of all affected registrations. The two functions must operate in a connected workflow, not in parallel silos.
- Deadline calendaring with ownership. Every renewal date, submission deadline, and fee payment window needs an assigned owner and a secondary point of contact. EUDAMED’s May 2026 deadline is a fixed date with no flexibility, and the internal QA readiness required before that date must be planned months in advance.
The FDA treats registration and listing as catalog maintenance, not a one-time event. That mindset is the right one. If your team still operates under the assumption that registration is completed at launch and revisited only when required, you are creating compounding risk with every product update and every market expansion.
Pro Tip: Subscribe to regulatory update feeds from FDA, EMA, and HSA directly, not through secondary summaries. Regulatory changes announced through those channels typically include implementation dates and transition guidance that secondary sources sometimes omit or mischaracterize.
For companies operating in Southeast Asia, aligning registration data with calibration and audit documentation is also worth attention. Inspectors reviewing GDP and GDPMDS compliance often pull registration records alongside distribution and storage documentation to assess whether the product being distributed matches the registered specifications.
My perspective on registration as an operational discipline
I’ve worked closely with healthcare companies navigating regulatory submissions across multiple markets, and the pattern I see most often is this: registration is treated as a project with a start and end date, managed by a single specialist, and parked until something forces a revisit. That model fails consistently.
What I’ve found actually works is treating product registration the same way you treat quality management. You build systems, assign ownership, set review cycles, and conduct internal audits. The EUDAMED implementation is a useful forcing function here. Companies that treated EUDAMED as a database to access in May 2026 are discovering that the real work is internal: aligning UDI data, coordinating role assignments, and reconciling labeling versions across markets.
In my experience, the teams that handle registration well are the ones where regulatory affairs and supply chain operate from the same data. When your pharma regulatory services team updates a registration, your logistics partner should know immediately, because import documentation, labeling compliance, and storage protocols often depend on that registered product status.
The advice I give consistently: build your compliance agility now. Regulatory demands across the US, EU, and Singapore are converging toward digital, real-time, and transparent systems. The companies that adapt their internal processes to match that trajectory will have measurably lower compliance costs and fewer market access disruptions over the next five years.
— Labgistics
How Labgistics supports registration and logistics compliance
Regulatory compliance does not stop at the point of registration. Maintaining market access requires that your product’s entire supply chain reflects its registered status. Labgistics provides specialized healthcare logistics solutions designed specifically for pharmaceutical and medical device companies managing complex compliance obligations across Southeast Asia.
From cold chain logistics and vendor-managed inventory to calibration services and regulatory support, Labgistics aligns operational infrastructure with the documentation standards that product registration and regulatory inspections demand. With over 20 years of experience supporting pharma and medical device companies in Singapore and across the region, Labgistics operates as an extension of your compliance team, not just a warehouse. Explore Labgistics’ pharma distribution services to understand how supply chain integrity and registration compliance connect in practice.
FAQ
What is product registration for pharmaceuticals and medical devices?
Product registration is the formal process by which pharmaceutical and medical device manufacturers obtain regulatory authorization to market their products in a given country. It involves submitting technical, safety, and efficacy documentation to the relevant regulatory authority, such as FDA, EMA, or HSA Singapore.
How do you register a medical device with the FDA?
FDA requires medical device manufacturers to complete both establishment registration and device listing through the FURLS system. Registration must occur within 30 days of beginning commercial distribution, with annual renewal completed between October 1 and December 31.
What is EUDAMED and why does it matter in 2026?
EUDAMED is the EU’s centralized database for medical device registration and market surveillance. Starting 28 May 2026, its first four modules become mandatory, requiring all economic operators to register actors, obtain an SRN, and submit UDI and device data before placing products on the EU market.
How does product registration in Singapore differ from FDA requirements?
Singapore’s HSA uses a risk-based classification system with multiple registration pathways, including abridged and verification routes for products already approved in reference markets. The 2026 updates introduced eCTD submission requirements and new GMP conformity assessments for overseas manufacturing sites.
What happens if product registration is not renewed on time?
For FDA-regulated devices, a lapsed registration can result in import detentions, misbranding enforcement letters, and suspension of commercial distribution rights. There is no grace period after the December 31 renewal deadline for US device establishments.